Test & Measurement
Welcome to Electronic Design's destination for test and measurement technology trends, products, industry news, new applications, articles and commentary from our contributing technical experts and the community.
Recent
Recent


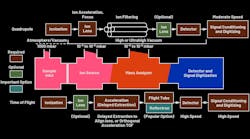
Keysight and Dreamstime_lescunliffe_23231826

Andrei Dzemidzenka, Dreamstime

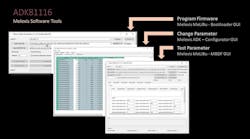
Dreamstime_ekkalucksangkla_1082822391

Highlights
Highlights
Electronic Design

Dreamstime_luchschen_26003212

Dreamstime_forfunlife_17805192
RIGOL Technologies

Dreamstime_marekuliasz_267841133


Recommended
Recommended

Rojdesign, Dreamstime
Warenemy_Dreamstime




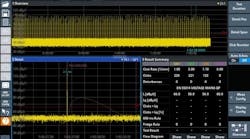
Anyaivanova, Dreamstime

Dreamstime_agsandrew_32581249
